

Imagine changing a baby’s diaper, setting it aside, and returning a short while later to find that the urine has turned a startling, jet-black color. Or picture hitting your thirties or forties and experiencing a form of arthritis so severe and rapidly progressive that it defies the typical aging process.
These are the real-world realities of alkaptonuria (AKU), a fascinating yet deeply challenging rare genetic condition. Often referred to colloquially as “black urine disease,” alkaptonuria is a classic example of an “inborn error of metabolism.”
While it is entirely non-infectious, its effects on the human body are profound, systemic, and progressive. Let’s dive deep into the science behind this condition, explore how it impacts daily life, and look at the modern medical solutions transforming patient care.
1. What is Alkaptonuria and why is it called that?
The name “alkaptonuria” sounds highly technical, but its origins are quite literal. The term blends Arabic and Greek roots: alkali (base), kapto (to suck up greedily or oxidize), and ouron (urine).
It was named because the urine of affected individuals possesses a unique chemical quirk: when it is exposed to air or mixed with an alkaline (basic) substance, it rapidly oxidizes and turns a dark brown or black color.
Historically, this condition holds a monumental place in medical history. In the early 20th century, British physician Sir Archibald Garrod studied alkaptonuria and used it to formulate the groundbreaking concept of “inborn errors of metabolism.” He realized that the disease followed a strict hereditary pattern, linking a specific genetic defect directly to a block in a metabolic pathway.
2. Is Alkaptonuria a rare disease and where is it most common?
Yes, alkaptonuria is classified as an ultra-rare genetic disease. In most parts of the world, it affects roughly 1 in 250,000 to 1 million people. Because it is so uncommon, many primary care doctors may go their entire careers without treating a single case.
However, genetics can cluster in fascinating ways due to geographic isolation and historical migration. Alkaptonuria is significantly more common in two specific areas of the world:
Slovakia
The Dominican Republic
In these regions, the prevalence rises dramatically to about 1 in 19,000 people. In Slovakia, this isn’t caused by one single mutation passed down through a family tree, but rather a specific cluster of multiple mutations within the local gene pool.
3. What is the cause of Alkaptonuria?
Alkaptonuria is an inherited, autosomal recessive genetic disorder.
To break that down into plain English:
Genetic: It is caused by a mutation in a specific gene—namely, the HGD gene.
Autosomal Recessive: A person must inherit two altered copies of the HGD gene (one from each parent) to develop the condition. If a person inherits only one mutated gene, they are a “carrier.” Carriers lead perfectly normal lives and do not show any symptoms of the disease.
Is Alkaptonuria a Bacterial Disease?
Because it involves changes in body fluid colors and progressive inflammation, people occasionally wonder if it’s an infectious process. To be absolutely clear: No, alkaptonuria is not a bacterial disease. It is entirely a genetic metabolic issue. You cannot catch it from someone else, and antibiotics have no role in treating it.
4. Which Enzyme is Deficient in Alkaptonuria?
The core culprit at the molecular level is a missing or defective enzyme called homogentisate 1,2-dioxygenase (HGD).
Under normal circumstances, your body uses enzymes to break down proteins from the food you eat. Specifically, the body processes two amino acids: phenylalanine and tyrosine.
The HGD enzyme is responsible for breaking down a molecular byproduct of this process called homogentisate (also known as homogentisic acid or HGA).
When the HGD enzyme is missing or functionally deficient, the metabolic conveyor belt grinds to a halt. The body can no longer break down HGA. As a result, this acid builds up in massive quantities in the bloodstream and tissues, leaking out into the urine and setting off a chain reaction of physical complications.
[Dietary Proteins] ➔ [Phenylalanine & Tyrosine] ➔ [Homogentisic Acid (HGA)]
⬇
❌ Missing HGD Enzyme ❌
⬇
[HGA Accumulates in Tissues]
5. What Happens to a Person with Alkaptonuria?
When homogentisic acid builds up in the body over decades, it doesn’t just sit there harmlessly. It undergoes a process of oxidation and polymerization, turning into a dark, pigment-like substance.
This dark pigment gets physically deposited into the body’s connective tissues—particularly cartilage, tendons, ligaments, and bones. This pathological staining and hardening of tissue is a medical phenomenon known as ochronosis.
As the pigment binds to cartilage, it alters the tissue’s structural integrity. The cartilage becomes brittle, weak, rigid, and highly prone to degradation. Over time, this leads to severe, structural damage throughout the skeletal and cardiovascular systems.
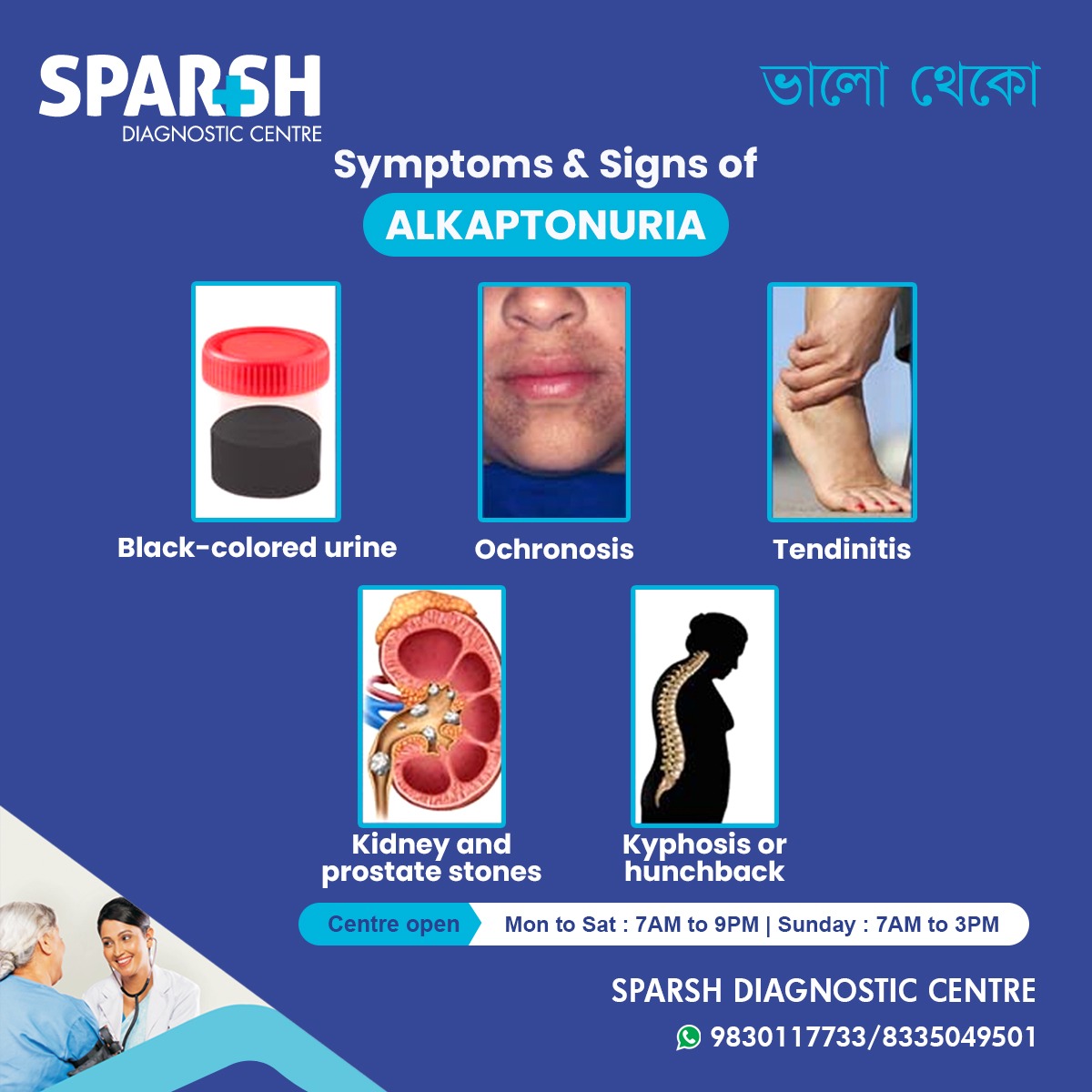
6. The Clinical “Triad” and Early Signs of Alkaptonuria
To identify alkaptonuria, clinicians look for a classic diagnostic hallmark known as the triad of alkaptonuria. This triad outlines the progressive narrative of the disease across a patient’s lifespan:
Homogentisic Aciduria: The excretion of homogentisic acid in the urine, causing it to turn dark upon standing or when exposed to alkaline environments.
Ochronosis: The bluish-black pigmentation of connective tissues (like the sclera of the eyes and the cartilage of the ears).
Ochronotic Arthritis: Severe, degenerative joint disease resulting from pigment deposition in large, weight-bearing joints and the spine.
What are the Early Signs?
The timeline of these symptoms is highly predictable. The absolute earliest sign occurs during infancy: dark stains in the diaper that become more pronounced if the diaper sits in an open trash can exposed to air.
Aside from the changing color of the urine, patients are typically completely asymptomatic throughout childhood and early adulthood. The deeper, structural signs don’t begin to emerge until a person reaches their late 20s or 30s.
7. Major Complications: Pain, Joints, and the Spine
Is Alkaptonuria Painful?
Yes, in its advanced stages, alkaptonuria can be highly painful. While the underlying metabolic glitch doesn’t hurt, the physical destruction of joints and cartilage certainly does.
As ochronosis advances, it targets the spine and large joints (like the hips, knees, and shoulders). The intervertebral discs in the spine flatten, calcify, and harden. This leads to chronic, debilitating back pain and a progressive loss of spinal flexibility.
Over time, many patients develop a distinct hunchback or a forward curvature of the spine, a condition medically known as kyphosis.
Beyond the spine, the tendons and ligaments throughout the body become brittle. A well-documented complication of alkaptonuria is the spontaneous tearing or rupture of major tendons—most notably the Achilles tendon in the heel.
Because the joint damage mimics aggressive osteoarthritis, it is incredibly severe; data shows that roughly 50% of patients require at least one major joint replacement surgery (such as a total hip or knee replacement) by the time they reach age 55.
8. Does Alkaptonuria Affect the Heart?
Yes, the heart is deeply impacted by the progression of alkaptonuria, and cardiovascular issues represent some of the most serious long-term risks of the condition.
The same black pigment that ruins joint cartilage accumulates in the connective tissues of the cardiovascular system. The primary targets are the heart valves, which are rich in collagen and cartilage-like tissue.
Valvular Heart Disease: The aortic and mitral valves are most commonly affected. As pigment deposits build up, these valves can harden, calcify, and narrow (stenosis), which restricts healthy blood flow. Alternatively, the valves may fail to close properly, leading to leakage (regurgitation).
Coronary Artery Disease: The pigment can also deposit within the inner lining of the blood vessels, accelerating the hardening of the coronary arteries (atherosclerosis) and increasing the risk of cardiovascular events.
Because of these risks, individuals diagnosed with alkaptonuria require regular cardiovascular monitoring, including echocardiograms, starting in mid-adulthood.
9. Other Complications: Stones and Systemic Build-up
Because the kidneys are working overtime to filter massive amounts of homogentisic acid out of the bloodstream, the urinary tract takes a heavy hit.
The high concentration of HGA and its metabolites frequently leads to the formation of hard mineral deposits. Patients with alkaptonuria have a remarkably high incidence of:
Prostate stones (in men)
These stones can cause severe episodes of acute pain (renal colic), urinary tract infections, and potential blockages that require surgical intervention.
10. How is Alkaptonuria diagnosed? Laboratory tests explained
If a doctor suspects alkaptonuria due to joint pain or dark urine, they rely on specific laboratory chemistry tests to confirm the diagnosis.
Why is Benedict’s Test Positive?
Benedict’s solution is a classic chemical reagent used to detect the presence of “reducing sugars” (like glucose) in a liquid. However, Benedict’s test is not entirely exclusive to sugars; it reacts to any strong reducing agent.
Because homogentisic acid is a powerful reducing agent, it strongly reacts with the copper ions in Benedict’s solution. When a patient’s urine containing HGA is heated with Benedict’s reagent, it yields a strongly positive result, typically producing a distinct color change and a dark precipitate.
The Ferric Chloride Test
Another quick, classic bedside check is the ferric chloride ($FeCl3$) test. To perform this test, a few drops of a ferric chloride solution are added directly to a fresh urine sample. If homogentisic acid is present, a transient, distinct deep blue or green color immediately flashes through the liquid.
While Benedict’s and ferric chloride tests are excellent historical and screening tools, the definitive “gold standard” for a modern medical diagnosis is a quantitative 24-hour urine test using Gas Chromatography-Mass Spectrometry (GC-MS). This test precisely measures the exact amount of homogentisic acid being excreted.
11. Modern Treatment Options and Management
For decades, medical professionals had very few tools to combat alkaptonuria. Treatment was almost entirely reactive, focusing on managing pain and replacing worn-out joints. Today, the management paradigm has evolved dramatically.
What is the Drug of Choice for Alkaptonuria?
The definitive drug of choice for treating alkaptonuria is nitisinone. Originally developed as a herbicide and later approved to treat a severe infant metabolic disorder called hereditary tyrosinemia type 1, nitisinone works by inhibiting an enzyme called 4-hydroxyphenylpyruvate dioxygenase.
By blocking this enzyme earlier in the metabolic pathway, nitisinone effectively puts a brake on the production of homogentisic acid. Clinical trials have demonstrated that low-dose oral nitisinone can reduce the levels of HGA in the urine and blood by up to 99%. This massive biochemical drop drastically slows down—or completely halts—the progression of ochronosis and joint destruction.
Why is Vitamin C Given?
Before nitisinone became widely available, and still occasionally used alongside other therapies, doctors frequently recommended high doses of Vitamin C (ascorbic acid).
Vitamin C is a potent antioxidant. It works by preventing or slowing down the oxidation of homogentisic acid into the dark polymeric pigment that embeds itself into tissues. While Vitamin C does not lower the actual amount of HGA being produced by the body, it can help reduce the rate at which tissue damage and staining occur.
12. Dietary Recommendations: What Foods are Good?
Because the toxic byproduct (HGA) comes directly from breaking down two specific amino acids—phenylalanine and tyrosine—dietary adjustment plays a supportive role in managing the disease.
A diet lower in protein can reduce the metabolic workload on the body. This is especially vital for patients taking nitisinone, as the drug causes a backlog that elevates tyrosine levels in the blood, which can sometimes cause eye irritation if left unchecked.
| Food Group | Food Items to Limit/Avoid (High Protein) | Safe, Preferred Options |
| Proteins | Red meat, poultry, fish, eggs, soy products | Strictly controlled portions of lean protein |
| Dairy | Hard cheeses, whole milk, Greek yogurt | Low-protein milk alternatives |
| Legumes & Nuts | Peanuts, almonds, lentils, chickpeas | Limited quantities of seeds |
| Grains | High-protein grains, quinoa | Rice, oats, standard wheat pasta |
| Fruits & Veggies | Minimal restrictions | Most fresh fruits and vegetables are excellent |
Note: Patients should never embark on an ultra-low protein diet without the close supervision of a metabolic dietitian, as your body still requires essential amino acids to function.
13. Prognosis: Is Alkaptonuria Life-Threatening and What is the Life Expectancy?
Living with a chronic, progressive genetic disease can be emotionally daunting, leading many patients to wonder about their ultimate prognosis.
Is Alkaptonuria Life-Threatening?
In and of itself, alkaptonuria is not considered an acutely life-threatening disease. It does not cause sudden organ failure or rapid terminal decline in youth.
What is the Life Expectancy?
Individuals with alkaptonuria generally enjoy a normal life expectancy. They can live well into their 70s, 80s, and beyond.
However, while it may not shorten your lifespan significantly, the condition profoundly impacts your healthspan—your quality of life during those years. The severe, chronic pain from ochronotic arthritis, the potential need for multiple joint replacements, the risk of tendon ruptures, and the developments of heart valve stenosis present major physical hurdles.
Fortunately, with early diagnosis, a managed low-protein diet, regular cardiac checkups, and modern therapeutic interventions like nitisinone, the modern outlook for individuals living with alkaptonuria is brighter and more manageable than ever before.
Frequently Asked Questions (FAQ)
What is the primary cause of alkaptonuria?
Alkaptonuria is caused by an autosomal recessive genetic mutation in the HGD gene. This mutation leads to a severe deficiency of the enzyme homogentisate 1,2-dioxygenase, preventing the body from breaking down a byproduct of protein metabolism called homogentisic acid (HGA).
Why does the urine turn black?
The urine turns black because it contains an abnormally high concentration of homogentisic acid. When the urine is voided and exposed to oxygen in the air, or if it comes into contact with an alkaline substance, the acid oxidizes and turns a dark brown or black color.
Can alkaptonuria be cured completely?
Currently, there is no permanent genetic cure to correct the underlying HGD gene mutation. However, the condition can be highly effectively managed with the medication nitisinone, dietary protein restrictions, and symptomatic care for joint and heart complications.
How does ochronosis affect the eyes and ears?
Ochronosis causes a slow buildup of dark pigment in connective tissues. In the eyes, this looks like small, slate-grey or black spots on the sclera (the white of the eye). In the ears, the cartilage hardens and takes on a visible blue-black or dusky discoloration through the skin.
Is it possible to have alkaptonuria and not know it until adulthood?
Absolutely. In fact, this is the classic presentation. Aside from dark stains in diapers during infancy, the majority of patients have no painful symptoms or obvious physical indicators throughout childhood. The severe symptoms, such as joint pain and ochronosis, typically do not appear until a person is between 20 and 40 years old.
To consult a Doctor or get full body check-up done at Sparsh Diagnostic Centre, call our helpline numbers 9830117733/ 8335049501.
#BhaloTheko
Disclaimer:
No content on this site, regardless of date, should ever be used as a substitute for direct medical advice from your doctor or other qualified clinician.
![]()


